Key FDA Policies, Procedures and Practices Before, During and After Medical Device Manufacturer Inspections

FDA medical device inspections are conducted by highly competent and dedicated medical device investigators. FDA managers and compliance officers provide an objective review of inspectional observations, Establishment Inspection Reports (EIRs) and corresponding manufacturer responses to inspectional observations, and determine if further agency action is necessary. FDA medical device investigators, managers and compliance officers follow detailed, comprehensive policies and procedures when conducting and reporting inspections, evaluating EIRs and determining post-inspection agency advisory, administrative or judicial actions when necessary. These policies and procedures assure the consistent application of FDA inspection and EIR evaluation processes no matter where in the world the inspection takes place.
Let’s take a closer look at the policies and procedures that shape your FDA medical device inspection.
Medical device investigators – training and competence

FDA investigators are typically hired with the education and experience necessary to develop in the commodity area they have been assigned (e.g. medical devices). FDA investigators complete basic training on the Federal Food, Drug, and Cosmetic Act and investigative interviewing and interrogation techniques. This training is supplemented by a curriculum of additional commodity specific training (e.g. online and on-the-job training experiences). These curricula are administered by FDA’s Office of Regulatory Affairs (ORA), Office of Training Education and Development referred to as ORA University (ORAU) and the investigator’s assigned division.
The Quality System Inspection Technique (aka QSIT, QSIT Guide or QSIT Manual) is the primary methodology for conducting routine FDA inspections of medical device manufacturers. QSIT is employed only by college-educated and competent FDA medical device investigators. According to Compliance Program (CP) 7382.845, Inspection of Medical Device Manufacturers, “Only QSIT trained individuals should perform these [medical device manufacturer] inspections.”1 According to the Investigations Operations Manual (IOM), “Inspections involving devices should be made only by those individuals qualified by training and experience in the device area.”2 This IOM statement expresses FDA’s expectation that, in addition to being formally trained, FDA investigators must be qualified by experience prior to being assigned complex, independent inspections of medical device manufacturers. This experience is typically gained by conducting team sample collections and inspections with experienced medical device investigators, attending advanced medical device training (e.g. process validation, industrial sterilization, etc.), and conducting independent inspections of progressively complex medical device quality management systems and medical device technologies.
Medical device investigators, compliance officers, supervisory investigators and management are taught and understand that they are the “eyes and ears” of the American public.3 These FDA professionals are the front lines for assisting in bringing new innovative devices to the consumer, for keeping effective medical devices available to the consumer and in assuring regulated manufacturers comply with applicable regulatory requirements. None of these obligations to the American consumer are taken lightly by FDA investigators. Acting primarily as scientists and engineers, FDA professionals engaged in the regulation of medical device manufacturers are trained to follow methods (e.g. IOM, CP 7382.845, QSIT) and base decisions on objective evidence (i.e. data supporting the existence or verity of something typically obtained through observation, measurement, test or other means).
Although FDA investigators frequently work alone, they are never without the support of their local management and a large network of subject matter experts (SMEs). If an investigator needs assistance or guidance during an inspection, they will reach out to a supervisory investigator or SME for guidance. SMEs include local specialist and national expert investigators as well as policy and technical experts located at ORA or the Center for Devices and Radiological Health (CDRH).
Preparation for inspections

According to its foreword, the Investigations Operations Manual (IOM) “is the primary operational guide for FDA employees who perform field investigational activities in support of the agency's public health mission. Accordingly, it directs the conduct of all fundamental field investigational activities. Adherence to [the IOM] is paramount to assure quality, consistency, and efficiency in field operations.”4
According to the IOM, “An establishment inspection is a careful, critical, official examination of a facility to determine its compliance with certain laws and regulations administered by FDA.”5 Moreover, FDA inspections are generally preannounced. Preannouncement of inspections was first introduced in the Federal Register.6 Section 5.2.1.1, Pre-Announcements, of the IOM states “Pre-announcements are mandatory for all medical device inspections in accordance with the criteria and instructions below....”7 The QSIT Manual also describes FDA’s policy of preannouncing medical device inspections.8 Preannouncement helps both the FDA and medical device manufacturers accomplish their objectives. Preannouncement helps the FDA with inspection efficiency by assuring the availability of appropriate records and personnel during the inspection. It also helps the manufacturer to understand the specific date, time and estimated duration of the inspection, the purpose of the inspection, the number of participating FDA personnel, the types of records and personnel to have available and, typically, the products and processes that will be inspected.
CP 7382.845, Inspection of Medical Device Manufacturers “provides guidance to FDA field and center staffs for the inspections and administrative/enforcement activities related to the Quality System (QS) Regulation (21 CFR Part 820), the Medical Device Reporting (MDR) Regulation (21 CFR Part 803), the Medical Device Tracking Regulation (21 CFR Part 821), the Corrections and Removals Regulation (21 CFR Part 806), and the Registration and Listing Regulation (21 CFR Part 807).”9
Establishment inspections (EIs) of medical device manufacturers may be conducted as statutorily obligated, per routine annual risk-based workplans, to assist in pre-market clearance or approval, “for cause” (or “directed”) to follow up signals identified through medical device reports or other means (e.g. trade complaints), for compliance follow-up or for other reasons.
According to CP 7382.845, inspections of medical device manufacturers “will assess the firm’s systems, methods, and procedures to ensure that the firm’s quality management system is effectively established (defined, documented and implemented) and effectively maintained.”10 CP 7382.845 states:
QS inspections should include the assessment of post-market information on distributed devices to include:
- Review of recalls
- Review of MDRs (Be alert to the fact that MDRs may contain information on recalls that have not been reported through the district under 21 CFR Part 806.)
- Review of corrections and removals
- Review of significant changes in device specifications or in the manufacturing specifications
- Follow-up on previous FDA 483 observation(s), to include the corrections, corrective actions or preventive actions for the observation(s) and the related system(s)11
According to CP 7382.845, “Available post-market information should be reviewed as a part of the preparation for the inspection, in order to facilitate efficient time spent at the facility. Identify in the EIR post-market information reviewed during the inspection, and adequately document your findings … Any problems identified as a result of the review of post-market information should be developed during the inspection.”12
FDA medical device investigators assess various post-market intelligence from a number of sources (including FDA’s Medical Device Reporting database, Manufacturer and User Facility Device Experience (MAUDE)) relative to a manufacturer and its products prior to conducting the on-site portion of the inspection. If any concerns are identified during the pre-inspection assessment, the concerns will be followed up during the on-site portion of the inspection. If, upon follow-up, the investigator identifies what (in his or her judgment) are believed to be objectionable conditions, these conditions will be communicated to the manufacturer during the inspection and at the conclusion of the inspection via discussions and issuance of an FDA 483 List of Inspectional Observations.13 The investigator’s observations will also be documented in the EIR representing the inspection for review by FDA management and potentially an FDA compliance officer.
Prior to an establishment inspection (EI), FDA investigators also review the manufacturer’s establishment file (EF). The EF contains previous EIRs, communications between the manufacturer and FDA (including responses to FDA 483 observations, promised corrections and corrective actions and evidence of implementation, labels and labeling, etc.), communications between the FDA field office and CDRH (including requests from CDRH to follow-up on signals identified during review of medical device reports, recalls, requests from the field office for annual reports, etc.) and other information to be followed up during the next EI (e.g. consumer or trade complaints). This information is used by the investigator during the inspection to focus on potentially problematic elements of the manufacturer’s quality management system.
Design, development and validation of the Quality System Inspection Technique
According to CP 7382.845, “QS [Quality System] inspections should generally be conducted using the Quality System Inspection Technique (QSIT).”14 The QSIT Manual describes QSIT, an FDA inspection model designed, developed and validated by SMEs from FDA’s ORA and CDRH.15 QSIT provides the guidance necessary for a competent medical device investigator to conduct a comprehensive and consistent inspection of a medical device manufacturer’s quality management system – no matter where in the world the inspection is conducted.
As demonstrated in the diagram (recreated from the QSIT Manual), QSIT breaks a manufacturer’s quality management system into four primary subsystems: management controls, design controls, corrective and preventive actions (CAPA), and production and process controls (P&PC).16 Three additional subsystems (facility and equipment controls, material controls, and records, documents and change controls) support the four primary subsystems. In addition, two of the primary subsystems (CAPA and P&PC) are supported by an additional four “satellite” subsystems (medical device reporting, reporting of corrections and removals, medical device tracking and sterilization process controls). The type of inspection (e.g. Level 1, Level 2, etc.) will dictate which subsystems are inspected during an inspection of a medical device manufacturer.
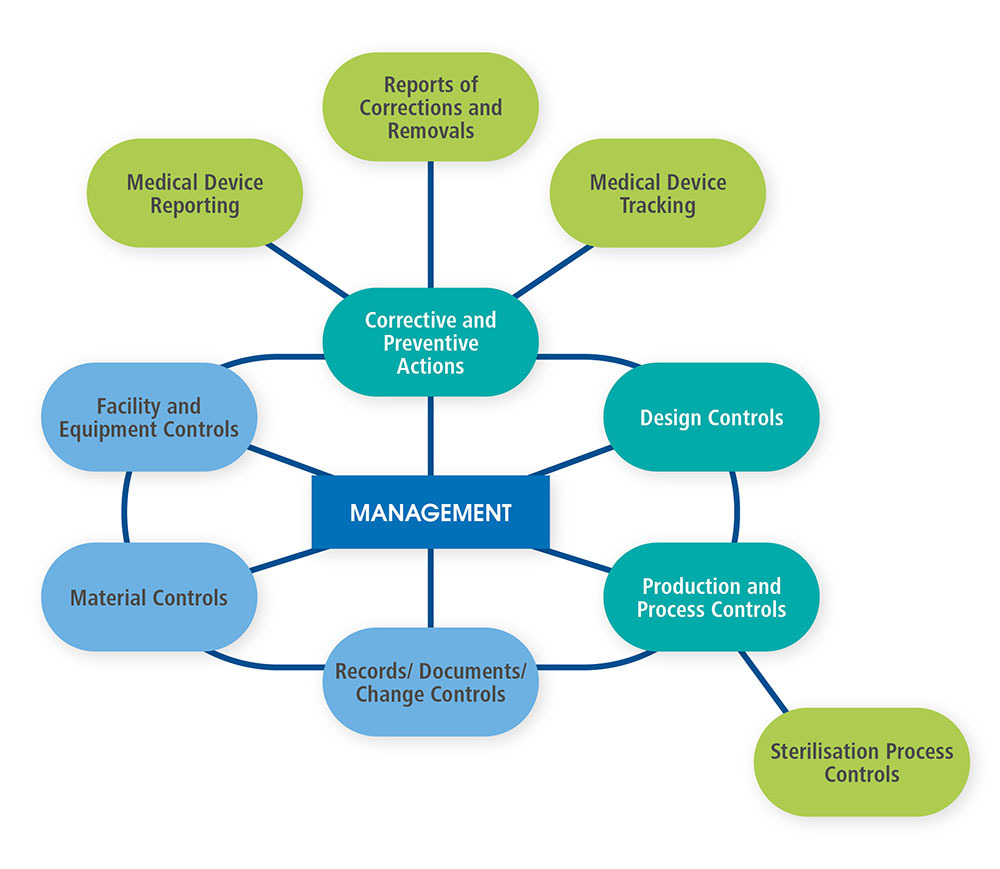
In describing the QSIT Manual, CP 7382.845 states “This QSIT tool can be scaled to meet the needs of each particular inspection.” The table below correlates the level of inspection and the guidance on how to perform the inspections.”17 CP 7382.845 includes the following table:
Inspection Level | Type of Inspection | Guide to Inspections |
|---|---|---|
| 1 | Abbreviated | QSIT – Two subsystems; Corrective and Preventive Actions (CAPA) plus Production and Process Controls (P&PC) or Design Controls (PAC 82845A) |
| 2 | Comprehensive | QSIT – The four major subsystems; Management Controls, Design Controls, CAPA and P&PC (PAC 82845B or 82845P or 82A800) |
| 3 | Compliance Follow-up | As directed by inspectional guidance and elements of QSIT (PAC 82845C) |
| Special | For Cause | As directed by inspectional guidance and elements of QSIT (PAC 82845G) |
| Special | Risk-Based Work Plan | As directed by CDRH inspection assignment and elements of QSIT (PAC 82845H) |
According to CP 7382.845, Level 2 inspections are conducted for initial inspections of Class III and, where possible, Class II manufacturers, by assignment, for foreign manufacturers, for training, for Accredited Persons inspections, when circumstances (described in CP 7382.845) necessitate a Level 1 inspection to be converted to Level 2 and, as district resources permit, for Class II and III devices.18
According to CP 7382.845, Level 1 inspections may be conducted for all routine surveillance and initial inspections of Class I and II devices, and for routine surveillance inspections for all classes of devices (I – III).19 Level 1 inspections are robust and focus on the most important elements of a manufacturer’s quality management system with respect to potential impact on device safety (i.e. freedom from unacceptable risk) or on identifying significant existing or potential nonconformities. The CAPA subsystem (including complaint handling and medical device reporting) will always be inspected. During a Level 1 inspection, investigators follow “linkages” into other non-mandated subsystems as appropriate. In addition to CAPA plus P&PC or design controls, Level 1 inspections often include the inspection of elements of other non-mandated quality system subsystems. FDA believes the inspection of CAPA plus either the P&PC or the design controls subsystem (with linkages to other subsystems as necessary) will provide a substantial review of the compliance status of the firm.20 Regardless of the type of inspection, the investigator will dedicate the time necessary to complete the objective of the inspection.
By review of an EIR, a reader can identify the type of inspection conducted by understanding the compliance program the investigator uses (e.g. CP 7382.845, Inspection of Medical Device Manufacturers) and the Program Assignment Code (PAC) cited. This information will typically appear at the beginning of the EIR. For example, if the EIR states that the inspection was conducted according to CP 7382.845, PAC 82845B, the inspection was a Level 2 inspection of a medical device manufacturer.21 A Level 2 inspection includes assessment of all four primary QSIT subsystems and their linkages to the remaining subsystems.22
According to CP 7382.845, a medical device manufacturer’s CAPA subsystem (including complaint handling and medical device reporting) is inspected by investigators during all routine Level 1 and Level 2 FDA inspections. This focus on CAPA activities will confirm that the manufacturer uses appropriate pre- and post-market intelligence to identify existing and potential product or quality problems, and that appropriate corrections, corrective actions or preventive actions are identified and implemented.
During design and development of QSIT, FDA employed contemporary design and development principles recognized throughout the world. For example, design validation is an essential element of good design and development principles. Design validation consists of test cases run under real-world or simulated use conditions. The objective of design validation is to confirm through objective evidence that the design (QSIT) meets user needs for its intended purpose.
FDA conducted a validation study of QSIT between 1 October 1998 and 18 February 1999. The QSIT design validation study involved a cross-section of investigators, managers and compliance officers from three FDA districts (Denver, Los Angeles and Minneapolis). During the QSIT validation study, a variety of manufacturers were inspected, from small to large multinational manufacturers and including low- to high-risk devices. Based on predetermined goals, empirical data was gathered throughout the QSIT validation study. The conclusion of this comprehensive study was that QSIT allows FDA investigators to conduct effective comprehensive inspections of medical device manufacturers using a defined and consistent methodology. A summary of the QSIT validation study and its results is found in the Medical Device and Diagnostic Industry article Validation of FDA’s New Quality System Inspection Technique.23
QSIT has been the primary inspection model employed by FDA medical device investigators to conduct inspections of medical device manufacturers worldwide since 01 January 2000 – a tribute to its design and development (attention to detail) and time-tested history of robustness, effectiveness and successful outcomes. The reliability of QSIT was demonstrated initially through empirical data derived from design verification and validation, and over 20 years of practical application during thousands of FDA inspections conducted worldwide.
Application of QSIT - example
According to CP 7382.845, inspections of medical device manufacturer quality management systems should generally be conducted using QSIT.24 Unless specifically exempted by regulation, FDA investigators have access to all documents and records required by regulation during an inspection of a medical device manufacturer. According to 21 CFR § 820.180, it is a regulatory requirement that such records be made readily available by the manufacturer for review and copying by the investigator.25 The following example shows how an FDA investigator applies the QSIT to assess a manufacturer’s compliance with medical device reporting requirements of 21 CFR Part 803.26
According to page 64 of the QSIT Manual, “The events described in Medical Device Reports (MDRs) may require the FDA to initiate corrective action to protect public health. Therefore, compliance with Medical Device Reporting must be verified to ensure that CDRH’s Surveillance Program receives both timely and accurate information.”27 The significance of thoroughly evaluating medical device reporting obligations of the manufacturer is clearly dictated to the investigator in QSIT (i.e. the investigator’s inspection instructions).28
QSIT provides specific medical device reporting inspectional objectives for the investigator to accomplish as well as narrative discussions relative to accomplishing those objectives. FDA investigators are obligated to accomplish each applicable QSIT inspectional objective including those relative to medical device reporting.
For example, according to the QSIT Manual, during their qualitative assessment of medical device reporting, investigators must:
- Verify that the firm has MDR procedures that address the requirements of 21 CFR Part 803.17
- Verify that the firm has established and maintains MDR event files that comply with 21 CFR Part 803.18
- Confirm that the appropriate MDR information is being identified, reviewed, reported, documented and filed
- Confirm that the firm follows their procedures and they are effective in identifying MDR reportable deaths, serious injuries and malfunctions29
FDA investigators are instructed to use one of two binomial sampling tables contained in the QSIT Guide to accomplish inspectional objectives relative to medical device reporting.30
The investigator’s selection of the appropriate table is primarily based on risk – either the risk of the device to the patient, user or others, or risk to the effective implementation of the quality management system. For example, Table 1 may be selected when inspecting a manufacturer of lower-risk devices, while Table 2 may be selected for the inspection of higher-risk device manufacturers. In my experience, potentially significant quality management system observations are often apparent to FDA investigators no matter which table is selected. This allows the observations to be identified independent of which table or sample size the investigator selects.
Investigators are not instructed to draw statistical conclusions based on the outcome of their sampling activities, and do not use the results of sampling to draw estimations or conclusions based on extrapolation.31 According to CP 7382.845, Part III A. 2. c.:
In addition, the QSIT sampling instructions allow the investigator to terminate a review of the sample when specific conditions exist (e.g. “You can terminate your review of the entire sample if you observe objectionable conditions beyond the number stated in the column header”).32 These instructions appear to reinforce the cited CP 7382.845 instructions with respect to the investigator avoiding data verification and analysis based on QSIT samples.
Post-Inspection activities
An FDA 483 is a “list of inspectional observations.” A cited observation does not represent a “violation” or inspectional conclusion, and does not mean that the company is not in substantial compliance with the regulations. If the investigator identified inspectional observations, an FDA 483 is issued to the manufacturer at the conclusion of an EI. The FDA 483 observations are, in the investigator’s judgment, potential violations of a regulatory or statutory requirement, as discussed in Section 5.2.3, Reports of Observations of the IOM:33
Section 5.2.7 of the IOM instructs investigators that during discussions of FDA 483 observations, the investigator must “Explain, in your judgment the conditions you observed may be determined by the FDA, after review of all the facts, to be violations.”34 [Emphasis added.] The IOM, Section 5.2.7, further instructs investigators that, during discussions of FDA 483 observations with management, “Do not assume the role of an authoritative consultant.”35 FDA investigators will not typically advise a manufacturer with respect to corrections and corrective actions relative to FDA 483 observations raised during an inspection.
The issuance of an FDA 483 is also not an enforcement action. FDA enforcement actions include warning letters, seizures, injunctions, criminal prosecution and criminal fines.36 Other actions available to the FDA are included in Field Management Directive (FMD) 86: Establishment Inspection Report Conclusions and Decisions, Section 6.5 Regulatory Actions (Advisory, Administrative, or Judicial).37 The FDA Regulatory Procedures Manual also discusses FDA advisory, administrative and judicial actions.38
The FDA 483 observations and supporting objective evidence collected during the inspection and documented within the EIR will be reviewed by FDA management and potentially a compliance officer (e.g. when advisory, administrative or judicial action is being considered).39 Typically, a manufacturer will respond in writing to FDA 483 observations providing specific responses to the observations, along with any proposed corrections and/or corrective actions (and any corresponding objective evidence of such). If received within a reasonable amount of time (15 work days) the manufacturer’s response will be considered by an FDA compliance officer prior to making any decision on advisory, administrative or judicial actions against the manufacturer.40 This process allows an independent reviewer (not the investigator) to make the final determination on any action taken in response to inspection findings.
According to CP 7382.845, Part V, at the conclusion of an FDA inspection of a medical device manufacturer, the EIR will be reviewed by a supervisory investigator or a compliance officer, and a compliance decision will be made.41
There are two compliance decisions available:
- “Situation I – The district has documented evidence indicating that one or more major deficiencies with the Quality System regulation have resulted in the inspection being classified as Official Action Indicated (OAI)”
- “Situation II – The inspection documents QS deficiencies of a quantity and/or type to conclude that there is minimal probability, in light of the relationship between quality system deficiencies observed and the particular device and manufacturing processes involved, that the establishment will produce nonconforming and/or defective finished devices. The Form FDA-483, Inspectional Observations, will serve to inform the establishment of any objectionable findings.”42
If the compliance decision is Situation I, the EIR will be classified Official Action Indicated (OAI). Official actions available to FDA include advisory actions (e.g. untitled letters and warning letters), administrative actions (e.g. administrative detention of devices and civil money penalties) and judicial actions (e.g. mandated recall, seizure of devices, injunction and criminal prosecution).43
If the compliance decision is Situation II, the EIR will be classified as either No Action Indicated (NAI) or Voluntary Action Indicated (VAI). In the case of a Situation II compliance decision (NAI or VAI), the manufacturer is expected to make voluntary corrections and corrective actions with no further FDA action necessary.44 Any corrections and corrective actions will be reviewed by FDA at the next scheduled inspection of the manufacturer. If the manufacturer fails to implement appropriate voluntary corrections and corrective actions, the next inspection may be classified OAI.
Receipt of the establishment Inspection Report by the manufacturer after an FDA inspection
Field Management Directive FMD-145 Release of Establishment Inspection Report (EIR) is FDA’s criteria and instructions for releasing a copy of the EIR to a manufacturer following the completion of an FDA medical device inspection and review of the EIR.45 According to FMD-145, “Inspections resulting in agency action (e.g., Warning Letter, Untitled Letter, Seizure) are not eligible for release until the inspection is closed in accordance with 21 C.F.R. § 20.64(d)(3). In the case of endorsed OAI inspections with Final Division Decisions, confirmation that no further agency action is planned from the appropriate Compliance Branch or Center must be obtained before release.”46 If the agency was contemplating an untitled letter, a warning letter, seizure, injunction, etc., the manufacturer would not receive a copy of the EIR under FMD-145. An FMD-145 copy of the EIR would only be released if the FDA considered the report to represent a compliance decision of Situation II (NAI or VAI). The receipt of an FDA-145 copy of the EIR by a manufacturer indicates the FDA is not considering further action as a result of the inspection.
Understanding the FDA policies, procedures and practices that shape your FDA inspection should help you understand what to expect prior to, during and after your next inspection.
How NSF can help
At NSF, we take a different approach. We focus on helping you build sustainable, lasting improvements at every stage of the product lifecycle, from regulatory compliance and quality systems to product development and beyond. With a team of seasoned regulators and industry professionals, we don’t just hand over solutions – we work alongside you to implement practical tools and strategies that keep your systems running smoothly. By empowering your team and strengthening your processes, we ensure that what we build together will continue to thrive long after we’re gone. It’s a true partnership, where your success is our success.
NSF’s range of consulting, training, and auditing services for medical device, IVD and combination product manufacturers are called upon by companies globally. Get in touch.
Learn more about NSF’s Medical Device & IVD Consulting, Auditing and Training Services.
Sources
1 FDA, Compliance Program Guidance Manual, Program 7382.845, Part II.B.5, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
2 FDA, Investigations Operations Manual, Section 5.6.1, available at https://www.fda.gov/media/113432/download (2020).
3 FDA, ORA Overview, available at https://www.fda.gov/about-fda/office-regulatory-affairs/ora-overview (“As the lead office for all FDA field activities, ORA serves as the eyes and ears of the agency ....”).
4 FDA, Investigations Operations Manual, Foreword, available at https://www.fda.gov/media/113432/download (2020).
5 FDA, Investigations Operations Manual, Section 5.1.2, available at https://www.fda.gov/media/76769/download (2020).
6 Federal Register, Vol. 61, No. 65 at 14789 (3 Apr. 1996), available at https://www.govinfo.gov/content/pkg/FR-1996-04-03/pdf/FR-1996-04-03.pdf; Federal Register, Vol. 63, No. 221 at 63933, available at https://www.govinfo.gov/content/pkg/FR-1998-11-17/pdf/FR-1998-11-17.pdf.
7 FDA, Investigations Operations Manual, Section 5.2.1.1, available at https://www.fda.gov/media/113432/download (2020).
8 FDA, Guide to Inspections of Quality Systems, available at https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf (Aug. 1999) (for example, page 13 of the QSIT Manual (“Preannounced Inspections”)).
9 FDA, Compliance Program Guidance Manual, Program 7382.845, Part I, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
10 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
11 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
12 FDA, Compliance Program Guidance Manual, Program 7382.845, Section III.A.1, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
13 FDA, Form 483 Frequently Asked Questions (9 Jan. 2020), available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/fda-form-483-frequently-asked-questions.
14 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1.a, available at https://www.fda.gov/media/80195/download (2 Feb.2011).
15 FDA, Guide to Inspections of Quality Systems, available at https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf (Aug. 1999).
16 FDA, Guide to Inspections of Quality Systems at 2, available at https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf (Aug. 1999) (coloring of graphic modified, content the same as shown in this report).
17 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1.a, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
18 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1.c, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
19 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1.b, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
20 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1.b, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
21 FDA, Compliance Program Guidance Manual, Program 7382.845 at 4 (“Field Reporting Requirements”) and Part III.A.1.b, available at https://www.fda.gov/media/80195/download (2 Feb. 2011) (referencing respective PACs based on type of inspection).
22 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1.a, available at https://www.fda.gov/media/80195/download (2 Feb. 2011) (table).
23 Medical Device and Diagnostic Industry, Validation of FDA's New Quality System Inspection Technique, available at https://www.mddionline.com/news/validation-fdas-new-quality-system-inspection-technique (1 Jan. 2000).
24 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.1.a, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
25 21 CFR § 820.180.
26 21 CFR Part 803.
27 FDA, Guide to Inspections of Quality Systems at 64, available at https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf (Aug. 1999). According to 21 CFR § 820.198(b), all complaints must be reviewed and evaluated to determine whether an investigation is necessary, and 21 CFR § 820.198(a)(3) describes that complaints must be evaluated to determine if the complaint represents an event which is required to be reported under 21 CFR Part 803 Medical Device Reporting. There is no regulatory obligation for manufacturers to thoroughly investigate all complaints received.
28 FDA, Guide to Inspections of Quality Systems at 62-66, available at https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf (Aug. 1999) (addressing Inspectional Objectives related to MDR, a flow chart, and further related discussion).
29 FDA, Guide to Inspections of Quality Systems at 64-66, available at https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf (Aug. 1999).
30 FDA, Guide to Inspections of Quality Systems at 104-107, available at https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf (Aug. 1999).
31 FDA, Compliance Program Guidance Manual, Program 7382.845, Part III.A.2.c, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
32 FDA, Guide to Inspections of Quality Systems at 104, available at https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf (Aug. 1999).
33 FDA, Investigations Operations Manual, Chapter 5, Section 5.2.3, available at https://www.fda.gov/media/113432/download (2020).
34 FDA, Investigations Operations Manual, Chapter 5, Section 5.2.7, available at https://www.fda.gov/media/113432/download (2020).
35 FDA, Investigations Operations Manual, Chapter 5, Section 5.2.7, available at https://www.fda.gov/media/113432/download (2020).
36 FDA, Types of FDA Enforcement Actions, available at https://www.fda.gov/animal-veterinary/resources-you/types-fda-enforcement-actions (6 Nov. 2017).
37 FDA, FMD 86: Establishment Inspection Report Conclusions and Decisions, Section 6.5, https://www.fda.gov/media/87643/download (28 Jan. 2014).
38 FDA, Regulatory Procedures Manual, Chapter 4 (Advisory Actions), Chapter 5 (Administrative Actions), and Chapter 6 (Judicial Actions), available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-manuals/regulatory-procedures-manual (applicable date depends on Chapter).
39 FDA, FMD 86: Establishment Inspection Report Conclusions and Decisions, Section 3.A.-B., https://www.fda.gov/media/87643/download (28 Jan. 2014).
40 Federal Register, Vol. 74, No. 153 at 40212, available at https://www.govinfo.gov/content/pkg/FR-2009-08-11/pdf/E9-19107.pdf (11 Aug. 2009).
41 FDA, Compliance Program Guidance Manual, Program 7382.845, Part V, available at https://www.fda.gov/media/80195/download (2 Feb. 2011); FDA, FMD 86: Establishment Inspection Report Conclusions and Decisions, Section 3.A., available at https://www.fda.gov/media/87643/download (28 Jan. 2014).
42 FDA, Compliance Program Guidance Manual, Program 7382.845, Part V.A.1.a.-b, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
43 FDA, Compliance Program Guidance Manual, Program 7382.845, Section V.A.1.a, available at https://www.fda.gov/media/80195/download (2 Feb. 2011).
44 FDA, FMD 86: Establishment Inspection Report Conclusions and Decisions, Sections 6.1.A. & 6.1.B.4., available at https://www.fda.gov/media/87643/download (28 Jan. 2014).
45 FDA, FMD-145 – Release of Establishment Inspection Report (EIR), available at https://www.fda.gov/media/83055/download (31 July 2019).
46 FDA, FMD-145 – Release of Establishment Inspection Report (EIR), Section 2, available at https://www.fda.gov/media/83055/download (31 July 2019).
How NSF Can Help You
Get in touch to find out how we can help you and your business thrive.

What’s New with NSF

NSF Granted Reauthorization as a CMMC Third-Party Assessment Organization
January 8, 2025
NSF Celebrates 50 Years of the Safe Drinking Water Act
December 16, 2024